Overview
The Ducho group’s main research interests are in the fields of
- Function-oriented natural product chemistry
Nucleoside antibiotics: synthesis, properties, biosynthesis Bacterial cell wall biosynthesis: intermediates, enzymology - Nucleic acid chemistry
Backbone modifications: synthesis, properties Nucleic acid labelling and functionalisation Nucleic acid delivery
Synthetic, SAR and biological studies on muraymycin nucleoside antibiotics
The main topic of our research is the investigation of nucleoside derivatives with potential antibiotic activity. Emerging resistances of bacterial strains, e.g. methicillin-resistant Staphylococcus aureus (MRSA), cause a significant need for novel antimicrobial agents. One approach in order to achieve this goal might be to perform systematic studies on naturally occurring antibiotics, particularly if they display novel or yet unexploited modes of action.
Muraymycins (e.g. muraymycins A1 1, A5 2 and C4 3, Fig. 1) belong to the class of nucleoside antibiotics and were isolated as a collection of 19 compounds from Streptomyces.[1] They inhibit the bacterial membrane protein MraY (translocase I), a key enzyme in the intracellular part of peptidoglycan biosynthesis, representing a novel target for antibacterial agents.[2,3]
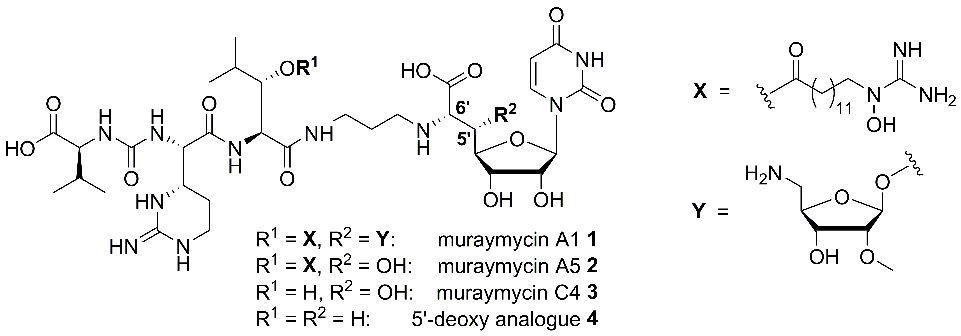
Muraymycins of the A-series showed the strongest antibacterial activities within the collection, namely against S. aureus, while non-lipidated analogues such as muraymycin C4 3 (i.e. compounds without a fatty acid moiety as residue R1, Fig. 1) were nearly inactive.[1] This strongly suggested an essential role of structural motifs of type X (Fig. 1) for cellular uptake[4] as the active site of MraY is located on the cytosolic side of the membrane.
We have developed synthetic routes towards the individual constituents of muraymycins, i.e. stereoselective syntheses of suitably protected building blocks, which were then connected in a modular fashion.[5-9] This also included nucleoside-derived building blocks for the preparation of 5′-deoxy analogues such as 4 (Fig. 1).[9 11] The 5′-deoxy scaffold was designed as a simplified variant of the muraymycin backbone because muraymycin A5 2 was only slightly less active than congener A5 1. Hence, the 5′-substituent was apparently not essential for MraY inhibition. We could indeed demonstrate that synthetically obtained 5′-deoxy muraymycin C4 4 was a reasonably potent inhibitor of MraY, which validated the concept to prepare 5′-deoxy analogues for further SAR studies. In addition, compound 4 also showed promising pharmacokinetic properties in vitro (sufficient stability in human plasma and metabolic stability) and was therefore identified as a novel lead structure for further development.[9] In addition to the 5′-deoxy scaffold, we have also developed efficient synthetic routes towards 5′-epi- and 6′-epi-muraymycins (structures not displayed).[12] It was therefore possible to synthesise a small collection of muraymycin analogues for biological testing.
For the biological evaluation of muraymycin analogues, we perform assays in cellulo (i.e. the determination of minimal inhibitory concentrations (MIC values) in bacterial cell culture) and also in vitro inhibition assays with heterologously expressed MraY.[13-16] These are complemented by model studies regarding the cellular uptake of muraymycins.[17,18]
Biosynthesis of muraymycins and related nucleoside antibiotics
As an additional project, we also conduct studies on the biosynthesis of muraymycin antibiotics and related natural products in collaboration with Prof. Steven Van Lanen, University of Kentucky, USA. We have been involved in the elucidation of the early stages of the biosynthesis of A-90289 antibiotics, which are sturcturally related to the muraymycins via their identical nucleoside core. The transaldolase LipK could be identified and characterised as the key enzyme for the formation of this uridine-derived moiety.[19] Based on the search for similar transaldolase genes in scarcely studied Actinomycetes, the sphaerimicins could be identified as a novel class of nucleoside antibiotics.[20] Similar transaldolase activity is also essential within the biosynthesis of capuramycin antibiotics, as studied based on a set of synthetic reference compounds which we provided the Van Lanen group with.[21]
Synthesis, properties and applications of modified oligonucleotides
The second main research project of the group deals with novel structural motifs for the synthesis of modified oligonucleotides (Fig. 2). The goal of this modification is the introduction of positive charges into the DNA backbone in order to study fundamental aspects of the role of the charge pattern in genetic molecules. Furthermore, such oligonucleotides can also be relevant for biomedical applications. For example, we envision the development of accordingly modified DNA-derived antisense oligonucleotides or antagonists of micro-RNA, with the modification leading to improved nuclease stability and possibly also to enhanced cellular uptake of the (partially) zwitterionic oligonucleotides. It is also anticipated that DNA analogues with a reduced number of negative charges in the backbone should be more amenable to conjugation with cationic cell-penetrating peptides. The presence of the reactive amino groups in the backbone will potentially enable further derivatisation and functionalisation.
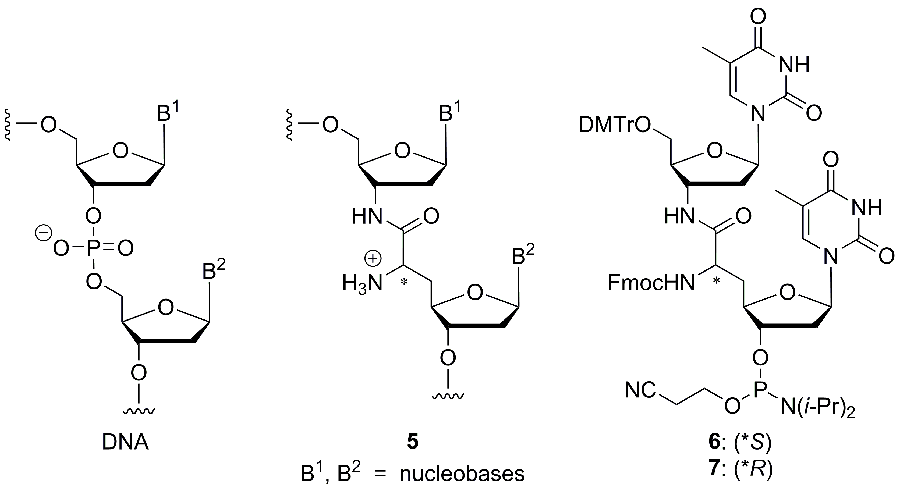
The novel ’nucleosyl amino acid (NAA)‘ modification of type 5, which has been inspired by the nucleoside structure of muraymycin antibiotics, was incorporated into oligonucleotides, following the synthesis of the phosphoramidite reagents 6 and 7 for automated DNA synthesis (Fig. 2).[22] Thus, dimeric T-T subunits could be replaced by the modified structure in a series of model oligonucleotides. This synthetic methodology was then extended towards the according replacement of A-T motifs.[23] The influence of the novel internucleotide linkage on the properties of the oligonucleotides, particularly on duplex stability, was studied in detail. We could demonstrate that NAA-modified oligonucleotides formed reasonably stable duplexes in particular with native DNA counterstrands, and that the selectivity in nucleobase recognition was not harmed by the presence of the cationic modification.[22] We therefore concluded that typical chemical properties of nucleic acids are retained in NAA-modified DNA oligonucleotides. Further studies on NAA-modified oligonucleotides are currently performed in our laboratories.
Literature
[1] L. A. McDonald, L. R. Barbieri, G. T. Carter, E. Lenoy, J. Lotvin, P. J. Petersen, M. M. Siegel, G. Singh, R. T. Williamson; J. Am. Chem. Soc. 2002, 124, 10260-10261.
[2] K.-I. Kimura, T. D. H. Bugg; Nat. Prod. Rep. 2003, 20, 252-273.
[3] M. Winn, R. J. M. Goss, K.-I. Kimura, T. D. H. Bugg; Nat. Prod. Rep. 2010, 27, 279-304.
[4] T. Tanino, B. Al-Dabbagh, D. Mengin-Lecreulx, A. Bouhss, H. Oyama, S. Ichikawa, A. Matsuda; J. Med. Chem. 2011, 54, 8421-8439.
[5] A. P. Spork, S. Koppermann, C. Ducho; Synlett 2009, 2503-2507.
[6] A. P. Spork, S. Koppermann, B. Dittrich, R. Herbst-Irmer, C. Ducho; Tetrahedron: Asymmetry 2010, 21, 763-766.
[7] M. Büschleb, M. Granitzka, D. Stalke, C. Ducho; Amino Acids 2012, 43, 2313-2328.
[8] O. Ries, M. Büschleb, M. Granitzka, D. Stalke, C. Ducho; Beilstein J. Org. Chem. 2014, 10, 1135-1142.
[9] A. P. Spork, M. Büschleb, O. Ries, D. Wiegmann, S. Boettcher, A. Mihalyi, T. D. H. Bugg, C. Ducho; Chem. Eur. J. 2014, 20, 15292-15297.
[10] A. P. Spork, C. Ducho; Org. Biomol. Chem. 2010, 8, 2323-2326.
[11] A. P. Spork, D. Wiegmann, M. Granitzka, D. Stalke, C. Ducho; J. Org. Chem. 2011, 76, 10083-10098.
[12] A. P. Spork, C. Ducho; Synlett 2013, 24, 343-346.
[13] P. E. Brandish, M. K. Burnham, J. T. Lonsdale, R. Southgate, M. Inukai, T. D. H. Bugg; J. Biol. Chem. 1996, 271, 7609-7614.
[14] P. E. Brandish, K.-I. Kimura, M. Inukai, R. Southgate, J. T. Lonsdale, T. D. H. Bugg; Antimicrob. Agents Chemother. 1996, 40, 1640-1644.
[15] T. Stachyra, C. Dini, P. Ferrari, A. Bouhss, J. van Heijernoort, D. Mengin-Lecreulx, D. Blanot, J. Biton, D. Le Beller; Antimicrob. Agents Chemother. 2004, 48, 897-902.
[16] A. Bouhss, M. Crouvoisier, D. Blanot, D. Mengin-Lecreulx; J. Biol. Chem. 2004, 279, 29974-29980.
[17] O. Ries, A. Ochmann, C. Ducho; Synthesis 2011, 2357-2368.
[18] O. Ries, C. Carnarius, C. Steinem, C. Ducho; Med. Chem. Commun. 2015, 6, 879-886.
[19] S. Barnard-Britson, X. Chi, K. Nonaka, A. P. Spork, N. Tibrewal, A. Goswami, P. Pahari, C. Ducho, J. Rohr, S. G. Van Lanen; J. Am. Chem. Soc. 2012, 134, 18514-18517.
[20] M. Funabashi, S. Baba, T. Takatsu, M. Kizuka, Y. Ohata, M. Tanaka, K. Nonaka, A. P. Spork, C. Ducho, W.-C. L. Chen, S. G. Van Lanen; Angew. Chem. 2013, 125, 11821-11825; Angew. Chem. Int. Ed. 2013, 52, 11607-11611.
[21] W. Cai, A. Goswami, Z. Yang, X. Liu, K. D. Green, S. Barnard-Britson, S. Baba, M. Funabashi, K. Nonaka, M. Sunkara, A. J. Morris, A. P. Spork, C. Ducho, S. Garneau-Tsodikova, J. S. Thorson, S. G. Van Lanen; J. Biol. Chem. 2015, 290, 13710-13724.
[22] B. Schmidtgall, A. P. Spork, F. Wachowius, C. Höbartner, C. Ducho; Chem. Commun. 2014, 50, 13742-13745.
[23] B. Schmidtgall, C. Höbartner, C. Ducho; Beilstein J. Org. Chem. 2015, 11, 50-60.